These pages cover subjects most of which I have talked about with the dozens of CF Patients and families who I have seen in consultation since The New Yorker article, "The Bell Curve". I have tried order the topics according to the frequency that these subjects have come up in the consultation conversations. I have added some new thoughts and observations that are related. If what you have questions about what is in these pages please ask me ‘Why?’ If I have left out a subject of subjects out that you would like me to write about please ask me by email at warwi001@umn.edu.
All are my thoughts based on my years of care of parents and families with CF. You should not consider that what I believe about how to treat CF is the "right" and the "only" way CF care should be given. There are many opinions of very good CF Doctors that are different from my opinions. I hope that your CF doctors will not be offended by any different opinion I have discussed with you or have written in the essay if what they do differs from what I do. If all doctors were to think the same then only one of us would be needed and we would never improve out treatments or improve on the future for patients with CF.
Remember the old English saying:
"Opinion between good men is knowledge in the making".
As your physicians disagree with some of the things that I have written; knowledge will be in the making.
I ask every patient, parent and spouse to tell me three good things about cystic fibrosis. Some have troubles but all rise to answer successfully. One mother took 4 days.
Examples of answers included:
I always add three of my own answers, based on observations I have made while caring for over 1000 Patients.
Cystic Fibrosis is the inheritance of a mutation in two CFTR genes, one each from both father and mother. These two inherited genes produce an increased risk of the person to acquire a large number of diseases. These diseases are common in the general population and include: bronchitis, pneumonia, sinusitis, nasal polyps, pancreatic insufficiency, diabetes, male infertility, and less frequently cirrhosis, symptomatic gallstones, and even kidney stones.
While this seems like a horrid number of unwanted problems there is a good side to this information. A person who has genetic cystic fibrosis and the CF Doctor know which specific illnesses he is at risk for and so will be able to take preventive precautions. Everybody has unknown genetic risk factors for other diseases but do not know what the risks or diseases are. So they have no idea of what actions might be taken to prevent their unknown potential problems.
I believe the name of this problem should be the original Cystic Fibrosis of the Pancreas because as preventive pulmonary treatments are attempted the percent of CFP Patients with the majority have pulmonary function tests in the normal range or above normal range. Over 97% have pancreatic insufficiency, over 70% have abnormal glucose tolerance tests, over 20% have diabetes, over 20% have two hour glucose levels characteristic of diabetes.
When I started working with cystic fibrosis the life table survival age was two years and now, at the Minnesota CF Center, the life table survival age is over 48 years. I believe the philosophy of prescribing the known preventive treatments to prevent as many of these problems as possible, the Minnesota's Cystic Fibrosis Center practice, is the reason for our Minnesota success. I am certain that the most effective approach is a vigorous airway clearance started at the time of diagnosis and continued twice daily. This is now built around our prescription of high frequency chest compression therapy.
There is another problem faced by young parents perhaps summed up as "What about having another child now that we have a child with cystic fibrosis." there is no shortage of unsolicited advice concerning what ought to be only a family question and decision.High Frequency Chest Compression (HFCC) systems manufactured today are the sine waveform Hill/Rom Model 104 and the ElectroMed SmartPulse plus the triangle waveform Respirtech InCourage System. The positive aspects of using HFCC therapy include: the simplicity of the HFCC technology, the ease of its use, and that it always works with 100% of the settings on the dials over the whole time of therapy. I believe that HFCC will always provide better treatment than other effective techniques including manual chest clearance, Autogenic Breathing and Active Cycle Breathing. My experience with the Flutter and Acapella is that they are seldom used well and so waste time, money and the opportunity for doing better airway clearance.
HFCC works most effectively when a patient inhales slowly and deeply. Exhalation should be passive. Always let the chest compressions oscillate the air in the airway passages while breathing through the nose. Patients should pause to cough after every five minutes of HFCC therapy. I recommend 2 daily HFCC therapy sessions of 30 minutes each for routine preventive therapy with an increase to 3 times a day during times when the patient has a worsening lung problem.
The sine waveform vests must be inflated before HFCC can start. This compression reduces the patient’s lung volume making breathing difficult. Then every patient must use extra energy and more muscles to take a breath. As the air in the chest increases; the volume of the vest decreases. That decrease in vest volume compresses the air in the vest which increases the pressure in the vest making it even harder to breathe.
HFCC works most effectively when a patient inhales slowly and deeply. Exhalation should be passive. Always let the chest compressions oscillate the air in the airway passages while breathing through the nose. Patients should pause to cough after every five minutes of HFCC therapy. With the Hill/Rom Models 103 and 104 and the MedPulse "sine waveform" machines the patient must remove a connection tubing from the vest, or the machine, in order to remove the chest restriction and so to be able to take the deep breath require for best coughing. The SmartPulse reduces the vest pressure to atmospheric when compression pulses stop. With the inCourage System "triangle waveform" machine the pressure drops to atmospheric when the vibrations stop and a tube does not need to be detached.
The Minnesota prescription for HFCC was developed over 16 years ago. It remains the model prescription for all HFCC machines. We found a wide scatter of ‘best’ velocities and volumes; sometimes with several frequencies having almost equal ‘best’ values. Every frequency was a best frequency for some Patients. For the 103 machine the largest volumes were under 10 Hertz (times per second) and highest velocities were usually over 15 Hertz. With the 102 machine both the best flows and volumes were less than 15 Hertz. The "triangle waveform" frequencies will be like the 102 machine. We shortened the therapy time from 60 to 30 minutes using the three highest velocities and the three largest volume frequencies each for 5 minutes and to save more time to do the Mucomyst aerosols during the 30 minutes of HFCC.
For the “102 machine I recommend frequencies 6, 7, 8, 10, 11 and 14.
For the InCourage System I recommend ramping frequencies from 5 to 15 and back to 5 under computer control over each five minutes period. However the 102 frequencies also work well.
Pressure and Frequency Interaction: There is an interaction between frequencies and pressure that has been recognized only after many years of HFCC therapy. Model 101 had only one pressure so no interaction could have been recognized. Model 102 had five pressures over a small range and most Patients did not know where to find the dial so that they never changed the pressure.
Model 103 had a visible dial for pressure calibrated 1 to 10 unlabeled units. It took us18 years of usage before we (Patients, parents, physicians and therapists) understood that a pressure that was comfortable at low frequencies could become intolerable at higher frequencies. When we found this out we developed a table for reducing the pressure when higher frequencies were used. (See chart below.) We have not worked out the details however we have worked out a guideline (See text below.) for Patients and parents to change the pressure and frequencies to find out which works best for each patient. We are confident that these principals will also work for Model 104 and the MedPulse and SmartPulse machines. For the InCourage System look after this discussion.
The best advice I have for a patient using Model 103 or any "sine waveform" system is to adjust frequencies and pressures using the following table. My preferred frequencies are 6, 8, 9, 18, 19 and 20.
| Frequencies to be used in sequence | Pressure Columns | |||||||
| 6 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
| 8 and 9 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 18, 19 and 20 | 1 | 2 | 3 | 4 | 5 | 5.5 | 6 | 7 |
For the "sine waveform" machines the pressure column is determined by starting with frequency 6 and pressure 3. Then after a few breaths increasing the pressure to 4 and if the patient notices no difference go to pressure 5…and so forth… until the patient notices that the new pressure changes the breathing, or makes it is harder to breath. That frequency is TOO high.
The prior lower pressure column is to be used to reduce the vest pressure at higher frequencies. For example if the patient notices a difference in breathing with pressure 7 then use pressure column 6 for all the frequencies. This will need to be checked twice a year for a growing child in good health and more frequently if changes in height or growth or health are significant.
I recommend that after every patient determine the pressure columns using the above directions for several weeks with each selection. Finally to use column 3, 2, 1 to find which of the three pressure columns works best. I will be very pleased to hear from all Patients about their experience and their finding the best to worst order of their three pressure columns. Send me an email at: <warwi001@umn.edu>.
The InCourage System uses a new valve to interrupt a powerful flow of air into triangle waveforms of energy. We did one clinical study to compare the power of this triangle waveform with the sine waveform produced by the Model 103. The triangle waveform using the frequency range for the square waveform machines and a comfortable pressure produced an average of 22% more sputum than the sine waveform delivered by the Model 103 using the table above with the difficult to breath limits.
Based on that I recommend that the InCourage System have a default program to start at frequency 5 Hz (pulses per second) and go up to 15 Hz and then back to 5 Hz over five minutes; then to pause for time to cough; then to repeat these two steps another five times. This ramping provides a virtual 3000 in sequence pulse rates in sequence from low to high and high to low over each 5 minute cycle. Thus all the airways regardless of dimensions will have optimum frequencies plus harmonics and hypo- harmonics many times throughout each cycle.
The inCourage System has a unique range of pressures. Pressures 10 to 30 have been designed for babies and small children. These pressures may also be useful for Patients who want to sleep using any frequency or a continuous ramping from 5 to 15 and back.. Although older Patients volunteer that they can breathe as easily with high pressures as with low pressures care should be taken because we have not yet found that there is no interaction between pressure and frequencies such as we have found in the widely used Model 103. It took 18 years with the 103 sine waveform to identify this problem because physician didn’t act upon children and adults reports and complaints. Eventually and by accident the discovery was made in my laboratory. If there will be another kind of frequencies and pressures I want to identify that within the first months the triangle waveform system is in use. So far we have found no suggestion that such an interaction exists with the inCourage System triangle waveform. Never-the-less I am soliciting reports from patient who has the InCourage System as they experiment with the pressures and frequencies out of their own curiosity or as directed by their physician or respiratory therapist. My email is warwi001@umn.edu.
Meanwhile, based on my experience with the sine waveform machines and the reports that each Patient can breathe easily with all pressures, I recommend keeping in mind that the triangle waveform delivers energy pulses with a higher impact with each pulse of energy. So if, Patients or Doctors or therapists, observe any interaction between pressures and frequencies that they would report their observations to me we so we can, in my laboratory, use their observations to improve my recommendations for use of the triangle waveform pulse technology. I will accept negative reports too..
The InCourage System has the same maximum pressure as Models 103 and 104 thousands of which have been useful for years even without my improved, but not finished, guidelines. The InCourage System has a wider range of pressures with the 10%, 20% and 30% settings being lower than any other HFCC machine in anticipation of the need for preventive HFCC starting in infancy. The pressure range 40% to 100% covers the range 1 to 10 in other HFCC machines. I recommend that most Patients start with pressure 40 and after each five minute ramp with a pause to cough several time to increase to the next pressure. Then if or when they find that the next pressure changes how they breath during the ramp that they use the prior pressure. Then each week to recheck the pressure is then same way.
The InCourage System jacket is designed to fit effectively when applied as directed.
I have not seen a satisfactory recommendation for the Models 103 and 104. Measure the greatest circumference of the chest (just under the arms or at the lowest part of the ribs above the abdomen). Adjust the standard vest to a circumference when adjusted that is 110% to 130% of that circumference. You will need a higher pressure with a greater vest circumference.
I do not have experience with the ElectroMed machines. However the suggestions for Models 103 and 104 may be helpful.
The principal of the guidelines we have developed for Model 103 and already discussed is a good way to start your personal experimentation to find the pressure(s) and frequency (ies) for you. If an increase of pressure alters your breathing or your breathing becomes uncomfortable assume that that pressure is, at least for a while, as too high for daily use. Keep in mind that as the airways become cleaner, as the InCourage System is used over a period of time, that a higher pressure may become more comfortable and more effective without a change in breathing. In general assume that Patients who are larger and so have larger jackets will probably need to use higher pressures. Some of the new Hill/Rom vests have such a large leak that the Patient can use the highest pressure and frequencies without effort or discomfort. We have not yet studied such leaky vests.
Our basic science research is largely done by Graduate Students in Electrical Engineering and supervised by Professor James Holte. They have developed much of the information discussed above and also confirmed earlier work.. They are studying the Minnesota HFCC technology that built the Models 102, 102, 103, 104 and the ICS systems. Their work so far has produced three MS and one PhD degrees. Their laboratory research ranking of estimated effectiveness, which will need to be confirmed by Patients and patient studies, is that the ICS > Models 101 and 102 > Model 103 > Model 104. An independent clinical study is under way to compare the ICS and the Model 104. Another one is needed to compare Model 103 and Model 104 because our unpublished laboratory studies suggest that the Model 103 is more effective than Model 104. I am aware of no published studies comparing any HFCC machines.
I am eager to hear of all observations or questions concerning any HFCC system because my laboratory developed the three waveforms and we are comprehensively studying Model 101, Model 102, Model 103, Model 104 and the InCourage System (we are not studying the MedPulse or the SmartPulse machines because the company that makes them will not give us machine or jackets for our research). Your observations will help us define tasks for the Graduate Students to solve. Their solutions to your observations or problems will improve all forms of HFCC. I reply to your observations or problems with whatever HFCC system you are using. Any suggestion or suggestions with concerning your use of our HFCC I will answer with the understanding that you will discuss my suggestions with your CF Doctor before you use them. You know yourself best, your CF Doctor knows best the medical and scientific problem that you have, so even if I know HFCC best, the best application of any suggestions I make must be worked out by you with your CF Doctor. My email address is: warwi001@umn.edu<.
There are times HFCC needs to be increased to three or more times:
1) The start of a cold. My regular recommendation is to start an extra therapy at the start of a viral respiratory illness and to continue the extra therapy for two weeks. The reason is that about 10 to 12 days after the start of a typical viral illness the body’s immune system takes a day or two rest after fighting the virus. During that couple of days a bacterial infection can get a head start and can cause a much more serious infection. The increased HFCC for two weeks will keep the airways less colonized by bacteria and clear out excessive bacterial growth until the immune system returns from that brief ‘rest’.
2) When a new lung infection or worsening of a chronic infection occurs. Then I prescribe three times a day HFCC with each HFCC increased time to 45 or 60 minutes. I favor a maximum of three times a day because there is less interruption of daily life with three than with four treatments a day.
HFCC treatments take at least 30 minutes twice a day so that leaves you only 23 hours a day to do things that you like to do, want to do or have to do. S0 small wonder that many patients find excuses to skip this health preserving treatment as often they can find an a plausible excuse. There is an option. Double tasking!
Double tasking has become a specialty for many people. My challenge for CF patients is to discover a second task to be done with their HFCC treatment. Then they will accomplish for each half hour a prescribed half hour of HFCC and a half hour of the chosen task that they like, want or have to do. The total for the day then is one hour used twice and so a 25 hour day has been achieved.
The risk of a person without CF of developing a serious lung infection today is about 0.01%. The chance of staying well of not catching serious lung infection today requiring antibiotics treatment is about 99.99%.
The risk of a person with CF, who does not do high frequency chest compression every day, of developing a serious lung infection today requiring antibiotics treatment, is about 0.5%. The chance of staying well and not catching serious lung infection today is about 99.5%.
The risk of a person with CF, who does high frequency chest compression every day, of developing a serious lung infection today requiring antibiotics treatment, is about 0.04%. The chance of staying well and not catching serious lung infection today is about 99.96%.
How these risks add up over a year is clear in this table:
| CF care | Today’s chance of being well | Today’s risk of being sick | Chance of being well this year | Chance of being sick at least once this year |
| No CF | 99.99% | 0.01% | 96% | 4% |
| CF no HFCC Rx | 99.5% | 0.5% | 16% | 84% |
| CF plus HFCC Rx | 99.97% | 0.03% | 90% | 10% |
Doing HFCC daily as prescribed can reduce the risk of a pulmonary infection 8.4 times as compared with not doing daily HFCC. A pay off, staying well, occurs about every six weeks.
Aerosol therapy has a long way to go before there is general agreement for use, time, medicines and equipment for therapy. I avoid all aerosols that provoke coughing by irritation. Although many Patients and physicians use hypertonic saline I prefer frequent coughing and the use of the Power Cough. Keep in mind that almost all Patients have a strong experience with controlling their cough. I prefer structured coughing as the best way to prevent spontaneous irritation and subconscious suppression of coughing.
I have prescribed the following aerosol during each vest therapy for over 30 years:
| Mucomyst 20% | 4 ml | |
| Albuterol unit dose (2 ml ampoule) | 1 vial | |
| Intal unit dose aerosol (2 ml ampoule) | 1 vial |
This yields 8 ml of a 10% solution of the Mucomyst which can be nebulized in less than 30 minutes with a good nebulizer. I recommend stopping the aerosol when the nebulizer begins to sputter. The mixture provides anti-inflammatory, mucolytic, bronchial dilation and mast cell stabilization. The 20% Mucomyst is concentrated enough to cause bronchospasm so that I dilute it to 10%. I use albuterol to assure dilated airways to improve the deep deposition of the Mucomyst and Intal to counteract the high probability (I estimate 40%) that allergies will interfere with good treatment.
The benefits of Mucomyst aerosols are offset by the odor which resembles rotten eggs, the stickiness of the not inhaled aerosol which can interfere with function of table top computers, television sets and other electronic equipment, and the risk of bacterial infection if the nebulizer is not carefully sterilized daily. Some of my Patients have substituted three times a day of:
One n-acetylcysteine (NAC) capsules size 500, 600, 800 or 1000 mg,
Two teaspoons of 10% solution of Mucomyst or
One teaspoon of 20% solution of Mucomyst.
Some Patients may still require use of Intal and/or albuterol by means of metered dose inhalers. Most do not need any inhalation medicine.
The Minnesota CF Center aggressive aerosol treatment component of preventive airway clearance protocol largely excludes Pulmozyme. Our most common observations indicate that Pulmozyme benefits only 1 in 8 Patients when they also use Mucomyst aerosols. Milla’s observations showed when Pulmozyme was added to the treatment of a hundred such Minnesota Patients, that these Patients had a more rapid drop of FEV1 than their matched Patients who did not use Pulmozyme (http://thorax.bmjjournals.com/cgi/reprint/53/12/1014. Suri, in The use of human deoxyribonuclease (rhDNase) in the management of cystic fibrosis. BioDrugs. 2005;19(3):135-44, reports that “the response to treatment is heterogeneous and only a proportion of Patients with CF actually benefit from the treatment. Rochat et al. suggests a mechanism for a rapid worsening when using Pulmozyme aerosols (http://erj.ersjournals.com/cgi/reprint/9/11/2200).
If your Doctor recommends Pulmozyme aerosols then these papers should be discussed with her. To be fair, on the positive side, Pulmozyme has a record of improving the lung function for many Patients when Patients have not had effective aerosol treatment and the Pulmozyme is compared to normal saline aerosols.
I believe that Pulmozyme may have some role in some Patients who are having rapid loss of lung function because I have observed three Patients who coincidently developed a rapid worsening of lung function preceding their deaths. I believe their rapid loss of pulmonary function can be aggravated by Pulmozyme aerosols. The infrequent benefit and my concerns about a rapid drop of lung function in a few Patients using Pulmozyme has led me to continue prescribing Pulmozyme only in the 10% of Patients for whom I can demonstrate improved lung function.
To be fair on the positive side, Pulmozyme is a wonderful enzyme designed to break the long molecules of DNA into very small pieces. A solution of DNA is very, very viscous. A solution of the short Pulmozyme broken fragments of DNA is very thin and runny. The first solution is very hard to move even with a cough; the second solution is so watery that almost any force, even gravity, will move it.
Consider what may happen when a patient with CF coughs with either solution in his airways. Without Pulmozyme the thick mucus will tend to adhere to the airway wall and the coughing may deform the cartilage and predispose to the development of bronchiectasis as the adjacent cartilage is injured. This may be a common injury when Patients not using Pulmozyme have coughing spasms and is the first reason why cough control needs to be learned by Patients and parents.
A coughing spasm with the thin and runny sputum when the cough does not stop will lead to an emptying of the air from the lungs that is so completely that the small airways are all closed. Then no more air can be expelled from the lungs and, despite efforts to breathe in or to cough no air can go in or out until the person can stop coughing and the closed airways can open. What happens, during that brief interval, is like what happens when a tooth paste tube is squeezed in the middle. Some toothpaste is applied to the tooth brush and some tooth paste goes deeper into the tube. I see a parallel event happening in the bronchi that are forced closed with the thin and runny liquid of DNA fragments; that watery sputum with bacteria is forced deeper into the airway branches, spreading the bacteria and leading to spread infection and loss of lung function. This may only occur in an occasional patient taking Pulmozyme and the associated rapid progression of loss of lung function is often looked as on unlucky event that has to be treated with antibiotics.
That coughing is important and good should be taught from diagnosis of CF. Even infants with no detectable lung disease need to be encouraged to cough frequently.. How this can be done is a problem that parents of infants and preschool children will need to help me figure out because physicians and physical therapist do not know how to teach children how to cough and to cough regularly in order to keep their airways free of excess mucus. This will have to be your experiment since no such studies have been described.
Every cough compresses the chest, narrows the airway diameters and forces the expiratory air to flow at higher frequencies. This accelerates the flow of mucus towards the mouth from which the sputum can be spit out or swallowed. Every patient who has the CF genes needs to develop a style of life in which each can cough many times a day in a way that their peers do not notice or ignore the frequent coughing.
All CF Patients need to learn to never suppress a cough. Cough suppression is a dangerous accomplishment. Not to cough in order to be socially acceptable is not acceptable. The goal is to learn how to cough frequently so no one notices the coughing.
The way to stop or to prevent a coughing spasm is simple, just hold one’s breath for a few seconds.
Another technique a patient can learn to prevent a coughing spasm can be learned. The technique is: First, learn to feel how much air is in the lungs at the end of a quiet breath, (the functional residual capacity (FRC), when all airways are normally open. Second, learn to fill the lungs to total lung capacity (TLC) by actively increasing the volume of the thorax. This will insure that all the alveoli behind each partly blocked airway will be filled full of air to push the mucus out with a cough.
The classic direction to fill the lungs is to tell the patient to take a deep, or big, breath. The quick breath that follows will the alveoli distal to the open airways and not the alveoli that are distal to the partially obstructed airways. The following cough will clear the clean airways well but not the partially obstructed airways. The better direction is "Fill your lungs".
Some Patients who have trouble with that changed direction may find filling the lungs by a series of regular inspiratory breaths with a pause without expiration after each regular inspiration. The lungs will be filled when no air can be inspirited by the last inspiration. Practice may be needed. When either method is performed well coughing is usually spontaneous. Even people with healthy lungs may cough when their lungs are completely filled with air.
This safe and effective cough from TLC and stopping at FRC should be done three times after each frequency or ramping cycle. This method of coughing achieves the goal of Autogenic Drainage while preserving the magnificent power of the cough to clean the airways. When using the Model 103, Model 104, the MedPulse and the SmartPulse machines disconnect a tube at the end of each frequency when the vibrations are stopped to remove the constant compression of the chest. The Model 101, Model 102 and the InCourage System have no background pressure since the pressure in their jackets become equal to room air pressure when the chest compressions stop.
There are many ways to use coughs instead of trying to suppress them. I have learned a few of them and am looking for more. My favorite cough is the magic cough. I learned the magic cough, from a magician, who has three children with CF. He told me that magicians let their audience see what they do but tricks the audience to see is what he wants them to see. For the magic cough, the patient uses a piece of Kleenex instead of a magician’s handkerchief. After coughing the patient takes the piece of Kleenex, blows his nose and spits into the Kleenex tissue while the Kleenex covers his nose. Everyone sees that the patient only blew his nose.
All my other coughs use similar obfuscations:
This technique was developed for use when HFCC cannot be done and to avoid extremes of coughing that may lead to coughing spasms that cause the airways to close and squeeze the sputum.
The movement of the squeezed sputum towards the mouth is beneficial. The movement towards the alveoli will distend and may damage distal airways and cause bronchiectasis or if the patient has sputum, thinned to watery consistency by dNASE, that sputum can spread an infection deeper into the distal airways.
This new technique uses active inflation of the lungs, air compression, passive exhalation of compressed air and passive inspiration to functional residual capacity. Coughs, described above, are used only when needed and then only after inflation of the lungs to total lung capacity. For Patients coughing up some blood or with postoperative pain this technique will clear mucus from the airways without coughing and decrease the risk of coughing up more blood.
Step 1. INFLATING THE LUNGS: Sit upright with the knees together, arch the back and stretch to be erect as hard as possible while lifting the shoulders and so spreading the ribs, rounding the chest and lifting the bony thorax. Then simultaneously, the yawning while trying to fill the lungs totally moving the diaphragm downward and so increasing the filling of the upper lobes). Alternatively the patient uses the "fill your lungs" or the breath step techniques described above.
Step 2. TRAPPING AND COMPRESSING THE INHALED AIR: The patient, after totally filling the lungs, closes the mouth and closes the nose with the fingers, slowly bends at the waist until the chest rests on the knees (This is done slowly so the air pressure difference in the alveoli can permit the flow of air through partially obstructed airways), The ribs come close together and more vertical in orientation and the abdominal contents push the diaphragm upward into the thorax. These actions reduce the lung volume towards residual volume while the compressed air keeps all the airways open. The hands are put on top of the head and the elbows are raised like wings or the arms are extended away from the body. Either adds the weight of the arms to aid compression of the air trapped in the lungs.
Step 3. PASSIVE EXPIRATION: The patient opens the mouth wide letting the compressed air out of the lungs. (Alternatively the hands may be placed behind the knees and the patient can pull the thorax against the legs for the first half of the expiration phase.) The patient does not cough. This decompression takes several seconds. The patient feels the air depart and the chest size decrease. The expiratory airflow velocity is amplified throughout the lung where the air moves through mucus coated narrowed airways creating greater ability to force the mucus toward the larger airways. The passive expiration position is maintained for a several seconds after the lungs feel empty to be sure that all airways empty and the lung has reached residual volume).
Step 4. RECOVERY: Returning to the starting position the patient slowly straightens up to the sitting position and lets the lungs fill with air without inspiring (The elastic recoil of the chest is sufficient to refill the lungs to FRC.) Once the lungs have filled the patient breathes normally.
Step 5. DECISION: The patient repeats steps 1-4 or goes to step 6.
Step 6. COUGH CLEARANCE. The patient fills the lungs to total lung capacity by either method and then coughs twice, quite hard. This cough does not go below FRC, the amount of air in the lung at the end of a normal breath. The cough from TLC has the greatest potential to move air through the airways. As long as the volume is equal to or greater than FRC the airways should be passively open so mucus plugs or watery mucus cannot be pushed deeper into the airways.
The process is repeated until the patient tires, feels the airways are cleared, or until a prescribed duration of the technique has been accomplished. When there is no electricity this should be done for the prescribed time for the HFCC therapy. This technique is not prescribed a number of repetitions because Patients invariably race to shorten the time for the therapy.
Although a full daily treatment should last for half an hour twice a day, a small number of repetitions done with maximal inspiration and with the rest of the steps can be repeated frequently during the day can be satisfyingly effective.
This technique requires only a chair and time. It can be done at anywhere at any time. While this technique is effective it shares the problem of all self-performed airway clearance techniques, i.e., it requires a dedication of time and close attention to technique. Perhaps it’s greatest potential is that it is simple, can be done anywhere and when there is no electricity for HFCC therapy.
Parents and Patients assume that role when they experiment with prescribed treatments and other directions. If they don’t talk with the CF Center Staff or learn their plans or discuss their variations of treatment then a major breakdown develops in what must be a joint fight against cystic fibrosis.
If the CF Center Staff does not ask about experimentation by Patients or fails to listen about an experiment it is hard to improve the treatment of the enemy, CF. If physicians, nurses, therapists, dieticians, etc. do not listen or recognize the importance of parents’ and Patients’ experiments, do not to pay attention to the experiments, do not try to understand the experiments do not find out what can be learned from the experiments and discuss how to improve future experiments there is a major breakdown in the joint battle against CF.
Doctors are slow to learn from Patients. Be patient with them and with the other staff in the CF Center. It took me many years to learn to ask Patients (and parents) what kind of experiments they are doing and to interpret their failures to comply with my directions as experiments rather than as their being non-compliant or non-adherent. Now I try to discuss the value of experiments and how to improve experiments. In particular I ask them to figure out what is a positive and a negative outcome and to discuss with me their ideas about an experiment before they do it, or, if they do the experiment on their own to share their plans and findings with me.
Doctors are slow to apply what they learn from Patients. I was fortunate to have
Annalisa Marzotto as my personal teacher during the years I served as her physician.
Annalisa determined that she knew more about herself and how my prescriptions and
directions worked if and when she used them and what she thought of them and how
she had decided they might be changed. My scheduled 40 minute clinic sessions
never lasted less than 90 minutes and often went 2 hours. And they occurred every
two weeks. Somehow Annalisa knew that Doctors seldom listen and she came
prepared to ask questions as well as to answer questions. Bu more important she had
answers to questions she wanted me to ask and she would use her answers as the
starting place for her questions such as
Why didn’t what you recommended work?
Why did what you recommended work differently that you expected?
Could it be because?
Could we try it this was?
Could we try this instead?
Was that idea based on wrong assumptions?
Would this be a better assumption?
How about this idea?
What do you expect your idea to do?
How will I know if your idea doesn’t work?
What do I do if your idea works differently than you expect?
Why did you not do?
Annalisa made these clinic visits very exciting and I looked forward to everyone. Annalisa changed me to be a collaborator with her with the goal to control the most serious complications of a CF patient that I have ever cared for. We worked together for over 15 years.
I’ve used many of the things she taught me with other Patients in a hap-hazard way over the years since she died. I never tried to make Annalisa’s teachings a principle for care of all my Patients until I have had the honor to being the consultant to many Patients and families who came to see me after reading "The Bell Curve" in the New Yorker. I have seen that all have needed to hear about Annalisa and her wisdom for the treatment of Cystic Fibrosis. As I consider Annalisa’s wisdom I believe her system of a Patient (or a family) and Doctor working together to treat the illnesses acquired because of the inherited CF genes is essential to living healthy and growing old with CF.
This will have to be learned by each Patient and family experimenting to learn how this approach can work with each Doctor -Patient association. I believe this may have to become a project for Patients and families to add to the Annual Family Education Days now part of all CF Centers annual education programs. The Doctors do not have the urgency to accomplish this because each probably has 70 or more patients. But each Patient has just the one doctor and so a one to one relationship must start with the Patient and the diagnosis of CF in order to be successful.
I know this is important but I do not know how to advise you. But I have enough ego to give it a novice try and to recommend that your successes be shared with other Patients and their families.
Parents and Patients need to read the medical literature, critically, and as co-partners in the fight against CF ask questions about what they read and learn. It is appropriate, from time to time, for them to read and explore the Internet and the medical literature critically and to ask questions about what they read and learn. Cutting edge science that exists in the Internet can be hard to find and more so to judge before a small part of the information becomes future corrected information. This is a formidable task for experienced professional CF Clinicians. Consider in 2004 there were 1,297 medical papers on CF with 2,080,200 Google hits. And if you add to you consideration a few of the CF associated diseases (diabetes, asthma, chronic obstructive lung disease, bowel obstructions and pneumothorax) there were 23,198 papers and 43,350,900 Google hits. No one has time to read more then a fraction of this much literature. Your doctor and your CF Center Team give you excellent care based on the medical literature of published reports all of which have been statistically vetted. These numbers show how hard it is for every CF Doctor to be up to date on every aspect of CF care. Because your findings and questions will be helpful and they should be well received.
Two reliable sources for generally correct information on cystic fibrosis treatment are:
http://www.nlm.nih.gov/medlineplus/ency/article/000107.htm and
http://www.cysticfibrosismedicine.com. Much of the commercial Internet data is
likely to be of uncertain reliability. Searching the Internet is like panning a river for
gold. Some streams have a lot of gold; some have only fool’s gold and others have
only sand.
Use the Internet with caution and with care.
This short list of other web sites of value is not complete:
www.cfcenter.umn.edu
www.cff.org
www.cForward.net
www.mycysticfibrosis.com
Read the fine print that comes with every prescription. You'll find that there are many side effects with every medicine. After you have a good understanding of your questions about CF, treatment variations, medicine in the literature and even non- prescription preparations you should meet with your (your child's) doctor and discuss your findings and your concerns. You and your (or your child’s) Doctor have to work together concerning each prescription, how to recognize if it is working and how to be aware of side effects. Every prescription is an experiment. The majority to times any given patient will respond the way the majority of Patients respond. The information on side effects and the interaction of the medicine with other medicines are also important for your benefit. Some of the side effects or interactions with other drugs are of little importance. Others may be significant. When you think a side effect or drug interaction could be significant you should discuss that with your (your child’s) Doctor.
You know your life style, your child’s life style, successes and failures very well. You need to bring this knowledge to every clinic visit and work out plans with the CF Center doctors and staff that you believe are good and that you can do as your part of the battle against cystic fibrosis. This is the time for you to protest if you cannot do what is recommended. If you don’t do this, you will leave all the decisions and experiments to your doctor who, as you know, does an experiment every time a prescription is written or a recommendation is made. Your doctor and your CF Center Team will give you excellent care based on the published reports, all of which have been statistically vetted, and their personal observations when these have been tried on other Patients.
Remember the published conclusions and recommendations have been formed using a Bell Curve like the one in the New Yorker article. Every Bell Curve has a 5 percent termination on each end and the middle 90% are considered to be normal. Remember that 1/3rd of published papers will be improved upon or proven wrong in the next five years. In every study some Patients will not have performed as well as the central "average" and some of the controls will have performed better than the central "average" that benefited. Be vigilant and talk with your CF Center and CF doctor.
Keep in mind that you and your doctor don’t speak the same language. Consider a couple of common errors that occur regularly:
When a doctor says he will "check on your child" in the hospital and when a patient says that he will "try to do what you recommend".
"I will check on your child (in the hospital)". The parents understand that the doctor said he will go to the hospital, examine their child, check the studies ordered etc. and do something. The doctor has committed to calling the hospital ward, talking to the doctor in charge about the child’s condition and laboratory studies and then to decide whether or not he should come in at once or wait.
"I will try to do the treatments you have recommended." The doctor hears that the parents and child will do what has been asked except for the occasional family crises that occur in any family. The parents perception is that have agreed to try the recommended tasks a few times and it they observe a difference they will continue but if they notice no benefit they will go back to what they have been doing.
Assumptions are dangerous. In either case further questions need to be asked to define what is meant so as not to conclude: "That doctor is not a man of his word" or "That patient or family is not compliant." Both doctor and patient have assumptions that lead to what they believe has been said and which can lead to disruption of future successful care of the CF acquired diseases.
Remember that the most dangerous assumption is that you know all that you need to know and that your CF Doctor has told you all that you need to know. As a member of the CF Care team for your child or for yourself you need to know all the details about prevention and treatment, how to wear and use the HFCC equipment, what the medicines do and don’t do and what are the risk factors and how to avoid them. Keep in mind that your CF Doctor knows a lot more that you probably appreciate but also does not know all the details that may be critical.
Consider how complex this can become when there is a problem of severe pain, a treatment problem for which few, if any, CF Doctors are experts. IN my experience of over 45 years I have participated in the care of perhaps a dozen CF Patients with severe pain that lasted over a few days. The problem of long term narcotic treatment to control severe I have seen in perhaps 5 Patients. For none of these patents have I been the primary CF Doctor but for all of them I have had great concern.
Your activities and participation are of equal importance with the activities, directions and prescriptions of the CF Center’s staff and physicians. The success of the CF Center is equally shared by the CF Patients and families and the CF Center Staff. You will need to work very hard to make this collaboration be successful.
One way to be ahead when your child with CF is young and is put to bed before you go to bed is to count the number of breaths you child take in one minute. Since every lung problem increases the respiratory rate the sleeping 60 second number of breaths can give you the earliest sign that your child is developing a lung infection. Exceptions are a dream or a full stomach. Count the sleeping respiratory rate (60 seconds) several times a week to discover the usual rate and the range of rates. Then when your child has a rapid rate assume that there is a lung problem or a dream. Count the rate again after half an hour and it the rate is still high do an extra HFCC treatment and call his CF Doctor if the respiratory rate is still high. This is the most reliable and earliest sign of an infection or other significant change in the lung function.
Consider ‘The Bell Curve’ since that title brought you to the Minnesota Cystic Fibrosis Center. That title was an over simplification since the studies considered only survival, height, weight, and pulmonary function tests. However many other ‘Bell Curves’ come with each patient starting CF care at each CF Center. Some of these ‘Bell Curves’ are age at diagnosis, the already acquired CF related diseases, their severity at time of diagnosis, the kind of CFTR gene mutations, the number of children in the family including other children with CF, sex, family income, existence and quality of the health insurance, one or two parents (each with no, one or two jobs), air pollution, distance from the CF Center, availability and kind and use of HFCC technology, knowledge of how to use the HFCC equipment, and so forth. Each such ‘Bell Curves’ affects each CF Center’s multiple ‘Bell Curves’.
You need to develop the communications skills that will place your role, as a patient or a parent, equal with your doctor's role, as a caregiver, with both of you fighting the clinical problem of cystic fibrosis. You will need to be able to explain to your doctor the changes in your body and in the function of your body and the response of your body to the prescriptions that are given. You need to develop enough of a background so that you can, with intelligence, discuss the treatments your doctor is prescribing. You’ll want to know the potential side effects from any prescription and the potential interaction between the different medicines that you are taking. For example; if you are taking multiple antibiotics: Are they to treat different bacteria? Are all needed to treat one bacterium? Should you be taking all of them continuously? If so, for how long? For example; if you are taking short and long acting bronchodilators, how can you determine that you need both kinds?
The battle against CF that you (as parent or patient) and your CF doctor and clinic work together to fight, is only part of the worldwide work, which in the United States is largely focused through the efforts of the Cystic Fibrosis Foundation (CFF). Help the local, regional and national efforts to raise funds to support the CFF’s planned programs. Remember the CFF supports your CF Center.
You and your CF Center’s doctors and staff have good ideas that need to be studied and smart questions that need to be asked concerning how to treat and how to understand the ways the risk factors for the CF associated diseases operate in your child’s and your CF Center’s unique environment. Discuss your questions with your CF Doctor and how you can help to do that research.
All CF parents and Patients need to be good observers, note keepers and reporters as they know themselves and problems better than anyone else. They need to keep notes of their observations and of questions so that, having them written down, when you see your CF Doctor you will not forget items that because they are important to you they are critical for your doctor to help solve your problems and to resolve your questions.
This may be difficult because likely your doctor has been given a fixed amount of time to see you, to answer your questions, to discover latent problems that may need attention, review your treatment and the results of your chronicle of changes in your treatment, to order new tests to improve his understandings, to make recommendations and write prescriptions. Most important to answer your questions about what you have learned about CF between your Center visits. You need to bring all of these to your clinic visit and work out plans with the CF Center physicians that you can believe are good and that you can do.
Since the pancreas injury often begins during pregnancy severely enough to produce meconium ileus is up to 10% of children with two CFTR mutations and pancreatic insufficiency before birth in about 95% if CF babies, nutrition problems are almost always a problem starting with their first feeding. So far that problem remains lifelong. For most Patients everything that follows is colored by the problems of digesting and absorbing the digested food and of a diversity of malnutrition problems which range from night blindness (Vitamin A deficiency), bleeding (vitamin K deficiency), osteoporosis (Vitamin D deficiency), failure to thrive and others well described in the CF literature. Pancreatic enzymes, extra fat soluble vitamins and essential fatty acid supplementations are needed, yet there are controversies concerning their necessities and amounts. Monitoring of these essential nutritional substances is necessary to avoid deficiencies. As excesses do not solve the problem of determining the optimum amount future studies must be planned. Meanwhile the subject of individual optimum amounts is one for Patients, parents and physicians to discuss.
Later digestive problems seen in some Patients include cirrhosis of the liver, gall stones, bowel obstruction, diarrhea, constipation, intussusception and gastro- esophageal reflux. These CF associated digestive problems response to the treatments for these problems in Patients who do not have CF.
I worry about using supplements when Patients are underweight. My position against "supplements" is that in almost all usage the supplement becomes replacement. Tube feeding I see as an occasional necessity which should be avoided if possible. Both gastro-esophageal reflux and inadequate or poorly used pancreatic can be causes of poor appetite and malnutrition.
Perhaps the most successful approach to malnutrition is the decision of the patient to eat more. the success because: "I decided to eat more." Do not underestimate the power of a decision even though you don’t know how to create the change in the patient’s brain.
There is a relationship between diabetes and nutrition. Diabetes is a constant worry before and after diagnosis. Avoid all liquids with sugar or high fructose corn syrup and to be wary of foods that have a high glycemic index. Fruit juice with meals may be a reasonable exception to liquids with sugar, but eating the whole fruit will be better. Restriction of high glycemic foods may be difficult when a patient with CF needs to gain weight. A concerned endocrinologist, preferably one who has or would like to develop expertise in CF related diabetes, may be needed in planning the diet for gaining weight. My concern about diabetes is real for already over 1/5th of our Patients have CF associated diabetes and almost another 1/5th have borderline diabetes. Another reason to avoid all soft drinks is the phosphate in these drinks can bind calcium and so increase the risk of osteoporosis.
I avoid fixed ratio vitamin preparations for Patients with CF such as ADEK or ABDEK, I believe that CF Patients need larger amounts of beta carotene, vitamin E, vitamin D, vitamin K, vitamin C, essential fatty acids, omega-3, vitamin B12, folic acid, selenium, with perhaps more supplements to be discovered. I recommend the serum levels of vitamins A, E, and D be checked at least once a year. Beta carotene, the precursor of vitamin A, is my preferable source for vitamin A since the body produces the optimum concentration of vitamin A it needs any excess is a useful antioxidant. A dietician can help you study these needs. You may find Roger J. Williams’s book, Biochemical Individuality, interesting. I have adopted his philosophical approach to optimum dose and nutritional side as the wave to consider the nutritional approach for CF.
I recommend that CF Patients try to maintain a weight 10% above average for age and height using the weight per height at age 18 as the index. The extra 10% should provide the muscles for good activity and body mass reserve in case of a serious infection.
Never-the-less and after years of constant efforts to gain weight gain some older Patients are having problems with obesity. This becomes a special problem because CF predisposes to diabetes and so does obesity. This needs to be recognized before or during adolescence so that obesity does not become another CF associated disease.
Exercise, especially aerobic exercise, is strongly advised throughout all of the European CF Centers. I have observed that CF Patients can do very well in all forms of exercise and that their health improves in proportion to their enthusiasm for both aerobic and strength building exercises. I recommend these exercises with supervision. Several of Minnesota CF Patients have received college scholarships because of their athletic successes in high school. Running sports are very good for people with CF. I have one patient who has run eight marathons. Soccer, cross- country racing and swimming are other good exercises in which CF Patients have excelled.
Nutrition should focus not only on calories but on water, quality of food and avoidance of sugar, high fructose corn syrup and high glycemic carbohydrates. Almost all food should be taken with enzymes. Even Patients who do not need enzymes for ordinary meals should take one or two capsules after eating a VERY large or VERY fatty meal to prevent acute pancreatitis.
For children, and even adults, who have normal stools and are not gaining weight an increase in the number of capsules with each meal will permit the patient’s subconscious monitor to permit him/her to eat more because with more enzymes the stomach will not be distressed in any way, for example having no discomfort or pain after eating.
It is an unwarranted assumption that there is a formula for the amount of enzymes to
be taken with a meal that is always right. The correct amount of enzymes is the
amount that will digest the food eaten and will vary with both the amount and the
composition of the food. My tests for determining the adequate amount of enzymes to
digest the food eaten are:
1) there is no stomach pain after the meal,
2) the stools sink,
3) there is no oil floating on the water,
4) passed gas is minimal and
5) not unusually foul smelling.
These tests define that the enzymes taken are appropriate for digestion of the food eaten. The last test is: "Is growth or weight adequate?" i.e. is the patient’s weight between 105 and 110 percent of expected weight for height. If that test is failed the slow increase in enzymes should be started and continued until ideal weight is achieved.
Glutathione is a combination of three amino acids: cysteine, glycine and glutamic. Cysteine is the active amino acid in this molecule. NAC is a more stable form of cysteine because it has only an acetic acid group attached. NAC is more water soluble and is the most cost effective way to increase glutathione levels in the body. N-acetylcysteine is sourer than glutathione but NAC has more antioxidant power. I recommend oral or aerosol n-acetylcysteine over glutathione. I have used NAC for aerosols for many years and now I am recommending Patients to try the oral route. NAC capsules are available and remove the problem of sour taste although their may be a faint odor. The cysteine amino acid part to both molecules has antioxidant, antiinflammatory and mucolytic actions. Glutathione, as well as NAC, has been given by aerosol and oral routes.
Hypertonic saline increases the water in the peri-ciliary fluid. It does this my attracting water to reduce the hypertonic saline to normal saline concentration. This extra water floats the mucus high enough that the cilia can beat and move the mucus out of the lungs. This activity lasts until the hypertonic saline is diluted to normal. I do not know how long this will take. A balancing consideration is that our studies have showed that the regular used of HFCC can increase the water content of water in the sputum for at least 12 hours after the last treatment. While the published studies show the effectiveness of hypertonic saline aerosols I prefer the HFCC method since it couples the increase of water on the mucus membranes with the removal of that mucus.
I believe that EVERY patient with CF has potential to improve lung function with improved airway clearance and with an optimistic approach to treatment, even those whose lung diseases have progressed to the point that they have been advised to consider a lung transplant. I have seen so many successful transplants that I support lung transplantation. However while there are many reasons for recommending a lung transplant, the only reason I favor is the desire is to improve the quality of life. When I recommend to Patients to consider the evaluation workup for lung transplant I advise these Patients to work as hard as possible to become so healthy that this option may be delayed both by themselves and by their physicians. I believe that many patients considering lung transplantation have the potential to improve their lung function with improved HFCC airway clearance and a determined and optimistic approach to treatment.
I recommend that all Patients with CF work hard and go to college to get a degree, and use that same effort and diligence to continue to work and to expect that a normal career is possible as well as marriage with children and even grandchildren to enjoy. For males, the classic CF infertility need no longer prevent fatherhood and, if not desired, adoption is a wonderful alternative. Every patient, or parent, should maintain a record of all clinical visits, lab work, x-ray reports, cultures, prescriptions and hospitalizations of the same quality as their CF Center Doctor’s medical records. Such records can be critical when traveling when seeking care from a new doctor or new clinic.
Every parent/patient should maintain a record of all hospitalizations, clinical visits, ab work, x-ray reports, cultures and prescriptions of the same quality as your CF doctor's medical records. Such records will be useful in many ways.
It is realistic to recommend that all Patients with CF should work hard and diligently to get a college degree. And after that, use the same effort and diligence to continue to work and to expect a normal career and along the way to marry and have children and even grandchildren to enjoy. For males, CF infertility need no longer prevent fatherhood and, if not possible, adoption is a wonderful alternative.
Birth and death are the mile stone markers of our lives. In between there is survival accompanied by health and accomplishments.
Death has always been linked with CF. From the early German song that a child that tastes of salt will die young to fund raising for a "cure". My early experience was being told by the doyenne pediatrician in Minnesota concerning babies with CF was "Warren, let them die. It’s easier for everyone."
The other option was to fight the death of children with CF by finding the cause of each infant and child’s death with over 400 autopsies. The result was that Minnesota became a Mecca of survival for children with CF. We organized and we were visited by CF doctors from around the world and that I was invited for a six week tour of the major cities in Australia to discuss cystic fibrosis. Our effort to create an international CF Database to use death as a marker of progress in survival collapsed when the CF Foundation removed it’s financial support. But countries around the world picked up the concept and almost every country has its own or participates in a CF Registry Database.
Our Minnesota CF Patient Care Database has data from diagnosis to date and to death. I questioned this dataset to find for every patient in the dataset for the year 2005 by year of birth, age at July first and the age of death for those who died. Then to use the age of death as a marker with which to determine survival of our patients for the year 2005 I calculated a Kaplan-Meier survival curve.
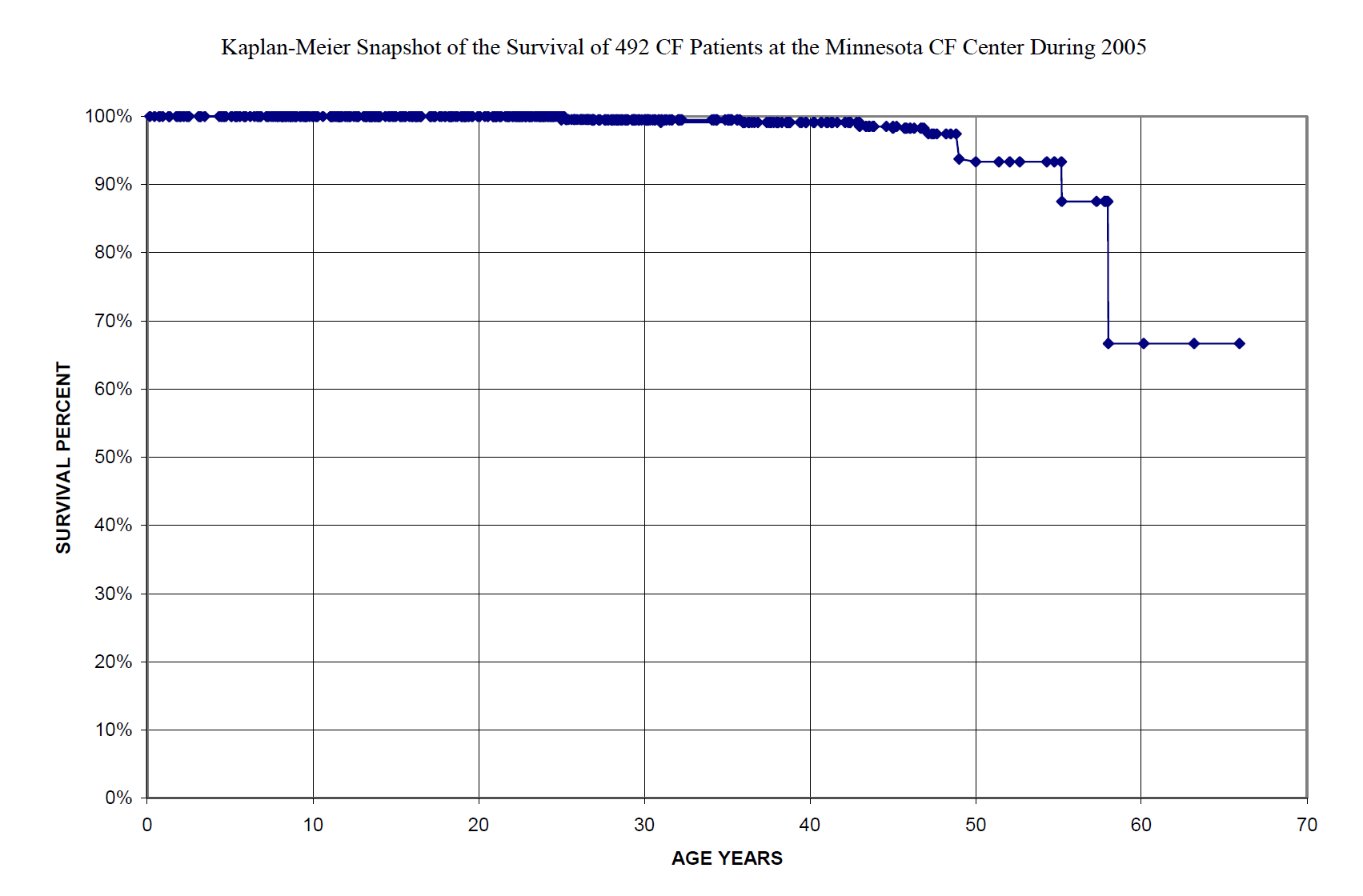
Each patient is marked by a diamond. Until about 33 years of age the diamonds are so close together they seem to be a solid line. Only after about age 48 do the numbers of patients decrease so that all, except for two alive at age 58, are represented as separate diamonds. The last diamond is at age 67 and percent survived of 67. This is not a hard scientific graph but is conforms to the requirement of Kaplan-Meier life table graphing. If you will give me my assumptions this is a true picture of CF patient survival in the Minnesota CF Center for the year 2005.
However this picture is incomplete in that it does not show details of health for that 29 of these survivors have had lung transplantation, that some had been diagnosed in middle age with ‘mild’ CF mutations, and that about 20% are handicapped by diabetes or advanced lung diseases.
MY THOUGHTS ABOUT LIVING WITH CYSTIC FIBROSIS have focused on the means of survival and preservation of health with CF that I have found to be helpful. I have tried to minimize the worry of every patients and family with death by telling all about our project to study aging in patients with CF in which we found that the longest survival so far with CF has been 83 years.
They have been limited without the consideration of why ‘death’ of each individual is important to the eventual normal life of current and future patients diagnosed as having CF genes.
Death is a mile stone and had been looked at in various ways, I like John Donne’s poem
DEATH be not proud, though some have called thee
Mighty and dreadfull, for, thou art not so,
For, those, whom thou think'st, thou dost overthrow,
Die not, poore death, nor yet canst thou kill me.
From rest and sleepe, which but thy pictures bee,
Much pleasure, then from thee, much more must flow,
And soonest our best men with thee doe goe,
Rest of their bones, and soules deliverie
Thou art slave to Fate, Chance, kings, and desperate men,
And dost with poyson, warre, and sicknesse dwell,
And poppie, or charmes can make us sleepe as well,
And better then thy stroake; why swell'st thou then
One short sleepe past, wee wake eternally,
And death shall be no more; death, thou shalt die.
All of us are engaged in a battle to beat the risks that cause the CF related diseases and that battle will continue for years. Patients and families need to remember that and if death happens then to continue the fight with a request for an autopsy and for the CF doctor to attend the autopsy. Every patient and family for whom I have cared for has felt that need to make the last contribution to beating CF. In over half of these autopsies we found new problems that we had not anticipated.
Now with childhood deaths almost vanished we have unknown problems in CF patients who die in middle and old age. No doctor, not even the physician who has cared for a patient for 20 or 30 years, can know all about the problems of patient’s health and causes of death and incomplete or even of missed diagnoses without an autopsy.
Not all the tools of medicine are advices, prescriptions, manipulations or surgery. The medical tools most needed are compassion and hope. Compassion in the doctor and patient relationship is where hope can be born and nourished. In the absence of compassion and hope there exists for the patient only despair.
The above comments have focused on the intellectual background for development of hope:
I see hope and despair as the extreme poles of a difficulty. Hope is the focus on a desired outcome; despair is the focus on an unwanted outcome.
The difficulty needs to be diagnosed and analyzed. Then options, all options, need to be identified and analyzed for such things as: cost, advantages, dangers and risks, how to apply, what may be accomplished, duration of benefit.
I’ve offered some of the ways I have analyzed CF and suggested approaches for my hope that most Patients with CF will, in my life time, achieve normal life expectancy. I’ve offered some of the ways I have analyzed CF and suggested approaches for my hope that most Patients with CF will, in my life time, achieve normal life expectancy. And yet I have concerns that many are dying prematurely and how to face death with hope. Beyond their faith I see another hope that may support both patient and family; the hope that they will have made a contribution through their fight against the CF acquired diseases from which they have suffered and the results of their autopsy to reveal still unknown answers to how better to fight their problems when such problems occur in other Patients with CF.
In the mean time I am working on high frequency body compression (HFBC) technology which applied both to the chest and the abdomen to oscillate all the abnormal functioning mucus membranes. I believe that all mucus membranes will have the same response to HFBC and so will change their cell membrane function so that their secretions will have normal water content. The triangle waveform oscillations are the most effective oscillatory form for producing this result. I believe that application of the triangle waveform oscillations to the mucus membranes in the pancreas, intestines, liver, and gallbladder soon after birth (or even during gestation) will allow these organs to grow without further obstruction and so to develop normal function during growth and then to normally throughout lives free of cystic fibrosis related diseases.
Whether my vision and hope for almost complete elimination of CF problems from the clinic and hospital will be the means for achieving this goal I am optimistic that that goal will be achieved because there are many other scientists and physicians who are also working with other approaches to achieve this.
We (Patients, families, husbands, wives, children, physicians, nurses, respiratory technicians, dieticians, secretaries, researchers, Foundations, and friends) can, working together, achieve this.
Practice hope.Good health and long life with cystic fibrosis depends on regular preventive care and a bit of luck. Success with attaining regular preventive care depends on the faith the patient and family have in their knowledge of CF and in the abilities of their CF Caregiver, on their hope that their use of preventive care will reduce CF risk factors close to normal and so increase chances of staying well. In becoming skilled in coping they will be in control of both preventive treatments and in their otherwise normal life.
Because the future course of CF is often set when the diagnosis of CF is made itshould be treated as a medical care emergency. The misinformation that exists in the community about CF makes even the suggestion that a child might have CF an occasion for panic for parents of a sick child. This distress when the possibility of CF is suggested, explains in large part the hesitancy of physicians to discuss the possibility with a family until they are virtually certain the child will have a positive sweat test. Such hesitancy may delay diagnosis so the patient comes to the CF Center with preventable complications.
The shorter the interval between the suggestion of the possibility of CF and the elimination or the confirmation of the diagnosis, the better it is for the mental health of the family and the better the opportunity for the CF physician to confront the family of the new patient with the paradox: that CF, the greatest genetic killer in childhood, is not a disease, that CE is only a genetic condition which makes the catching of several diseases that occur because of the paucity of water in the secretions of exocrine glands (mucus secreting cells and glands plus the sweat glands), and that the risk of catching these CF associated diseases can be reduced to near the risk of patients who do not have two CF genetic mutations with daily preventive treatments. The result can be a normal life.
The first hours after the diagnosis should be used to form a bond, which will foster the ability of the patient and family to work with the child's CF Doctor to achieve a normal life. The first contact must leave the family with several concepts secure in their minds. That CF is inherited by two autosomal recessive genes and that CF is not their fault. That untreated CF retains the potential for early illness or death that CF has always had, but that preventive therapy changes the prognosis. The parents should understand the difference between genetic predisposition and acquired complications because in this genetic disorder it is the acquired complications that cause clinical illness and in the past has frequently lead to death. The family with a child with CF needs to understand that much research needs to be done, that much of the information in the literature is all old and that the best source of new information about pathology, physiology, treatment and expectations will come through their regular follow-up clinic visits at their CF center. Over the years all contacts with the patient should reinforce these concepts.
Since the development of any complication in CF is a haphazard process influenced by medical and parental and patient behavior, the approach to the use of preventive treatments and simultaneously to practice a normal daily life is the major determinant of outcome. In facing a future of preventive therapy, hope can provide the energy to sustain the continuation of therapy in times of discouragement, weariness and stress. One of the great builders of hope is the continuity of care that exists in the CF Centers. The parents of the newly diagnosed child look at the teenager and hope that their child can be that healthy when the child reaches teenage. The teenagers look around and see patients in their twenties getting married and hoping that they will be as well when they graduate from college and look for a mate. Patients in their twenties seeinge patient in the forties who is physically active maintaining a full time job and hope to be that way when forty. Patients in their forties can see the patients retiring from their jobs and plan ahead for their days to collect social security. At all of stages the patients and their parents can look back and see what hope and help they are to younger patients and their families and be renewed in their own efforts to prevent CF complications and to live normal lives.
When the patients and families are in control of the preventive treatment they can be in control of making life and activities normal instead of letting CF control their lives. Patients, parents and CF Centers need a series of programs at all ages and with or without disease complications to develop skills to control their treatments and their lives so not have their lives controlled by CF complications or treatment.
First, behavior education incorporated into the initial training program for the families of newly diagnosed infants and young children concerning the physiology, pathology, pharmacology and techniques and goals of treatment. Second, a program of teaching coping skills using computerized teaching methods aimed at families of preschool and grade school children. Third, an annual Family Day to provide a forum for testing of social skills and sharing of accomplishments as well as problems. Fourth, a summer camp with other normal children to learn transition from parental care to self care for teenage patients. Fifth, how to integrate all aspects of CF of self care at home using the parents for supervision and back up to prepare for living in a dormitory with other college students who have their own health care problems who also need to adjust to the other social and psychological issues, i.e., dating, marriage, jobs. Sixth, additional interventions need to be implemented for patients who are handicapped by the acquired complications to develop self care and independent living skills.
The successful patient and family with CF are those who have faced CF recognizing that it is an inherited risk factor that can lead to early diseases and at the same time learn to compartmentalize the hours of the day so that the treatment of CF can be double tasked and intergraded with the normal activities of daily living, school, work, marriage etc. Patients are best able to accomplish this if they have a partnership with their CF caregiver in fighting CF together, as complete a knowledge as they can learn of all aspects of the CF gene mutations and the risk for CF related diseases, confidence in the efficacy of prescribed treatments and their personal ability to control the treatments to achieve the goals for their life. The knowledge and skill needed by the CF patient and family must be reviewed with every clinic visit and taught through problem oriented progression in sequence from diagnosis, throughout growth to adulthood and to old age. At every clinic visit the doctor sees a new patient and the patient sees a new doctor. Both bring new knowledge, problems and questions to share and to use together to fight and beat CF. Working together they can be successful.
After the amazing numbers of Patients, parents, friends and colleagues who came to the Gala for the awarding of the ‘Angela Warner Friend of the Foundation Award’ for lifetime accomplishments, I feel the need to tell you what I did not have time to say at the CF Gala. That there are "good" things about CF for Patients and families as well as for CF Caregivers and Researchers, and all of mankind. I want to tell you of good things I have seen over my 43 years of caring for children and adults with cystic fibrosis.
Rachael Kramlinger, a Minnesota CF Center patient, wrote about "Good Things" in a poster about CF she prepared for a school project. This is what she said that delighted and inspired me.
Inspired by Rachael, I reviewed my experience with CF and came to the conclusion that all children with CF are from Lake Woebegon. They "are above average"; they are almost always above average in intelligence and when we first test their lung function they are almost always superior in lung function. They often use intelligence to do well in school, college, graduate school, and professional schools. However, they are also normal and sneak out of doing their vest treatments or skip taking some of their medicines and so lung injury and disease can develop and may progress to being, in part, irreversible.
My personal experience with good things about CF as a caregiver, researcher and teacher is that●CF, as a medical problem, has been a great teacher for parents, relatives, friends, nurses, doctors, therapists, and scientists. As CF has changed over the years CF has continued to surprise us with new understanding and teachings of great value. Consider the few years we have known CF.
●Before 1950 CF was known as a rare and fatal gastrointestinal disease of infants and young children. In Medical Schools and teaching hospitals babies with CF filled a special place in the education of medical students and pediatricians. Interns and residents would comb the wards for rare diseases like CF to test visiting professors’ skills and knowledge. As medical understanding of CF developed during the late 1950s CF became recognized as a common genetic disease, perhaps as common as 1 in 1000 live births, and with some survivors into childhood and a very rare patient surviving into the teenage years.
●CF provided some real opportunities for CF and Genetic researchers. How could a fatal recessive genetic disease be so common; one child with CF in every 1000 births when almost all recessive genetic diseases occur in 1/10,000 to 1/30,000 births?
We learned three possibilities. First, families in the kindred in which CF occurred tended to have more children than the average family. Second, CF seemed to be more common in countries where, in medieval days, seafaring was a way of life. The explanation was that the increased salt in sweat of CF gene carriers, removing excess salt from the body, improved survival of sailors who had to drink salty water on long trips at sea. Third, the good immune system associated with CF may have improved survival in people who caught some of the epidemic diseases that killed large fractions of the European population.
●But, aside from such interesting intellectual problems, the early 1960s were a grim time. In that time the average survival age was 2 years. A child who survived more than the average often had 14 or more hospitalizations, sometimes up to four a year, before dying from nutritional, liver or lung complications. In thinking about the `60s one has to look hard to discover any good thing about CF. I recalled the medical chart of a beautiful little girl I cared for and lost. I reviewed her chart to refresh my memories of the first days of the Minnesota CF Center and discovered again the dismay and pain parents and children suffered. I recalled the advice the lead Pediatrician practicing in Minneapolis gave me, "Warren, just let them die. It’s easier for everyone."
I felt again the despair that led the CF Chapter to have special Christmas programs where CF children were showered with presents to distract them from their suffering. I relived my arguments with the CF Chapter opposing a special camp for CF Children. Our compromise: we arranged for the CF children to camp at Camp Courage, the camp for handicapped children. From that we learned the first good thing about CF from the CF Children. Despite having to sleep outdoors in camp tents turned into ‘mist tents’ they came back after seeing the handicaps other children had, saying, "I’m glad I have CF".
These CF Patients taught us the joy of taking advantage of the good times they had by radiating their joy when they felt well. Henrietta, my wife, helped me understand this from one of Gerald Hopkins’ poems:
All things counter, original, spare, strange; Whatever is fickle, freckled (who knows how?) With swift, slow; sweet, sour; adazzle, dim; He fathers-forth whose beauty is past change: Praise him.
We saw beautiful and brave children relishing each day despite discomforts of pain and breathlessness … and so they taught all who cared for them, their families and others that death awaits us all and that we should, with Moses, number our days and apply our hearts to wisdom.
●We observed a CF paradox, that survival with this fatal gastrointestinal disease was mostly dependent on acquired pulmonary infections. So, CF taught us the need to be flexible and creative in our thinking. The first Pediatric Pulmonologists were Pediatric Gastroenterologists who taught themselves, and then other Pediatricians, how to take care of and how to prevent or delay acute and chronic lung diseases in children. LeRoy Matthews, George Polgar, Will Waring and I organized a four- year program of across the country meetings educating Pediatricians about the pulmonary problems of childhood. We lobbied successfully for the creation of ten Demonstration Centers for the treatment of Pediatric Lung Disease. These activities led to the creation of Pediatric Pulmonology as a specialty and of a new medical journal with the same name.
●CF also taught us that CF was caused by an autosomal (non-sex chromosome) gene.
●CF also taught the need for the many other Medical Specialists to help treat CF and CF-associated diseases; and that all these caregivers would have to work together as a team.
●The National Cystic Fibrosis Research Association spawned the CF Club and then the CF Center Directors Group. The NCFRA and the CF Club permitted the Minnesota CF Center to create the CF Patient Database.
●As CF children survived and some new Patients were diagnosed in grade school or high school, CF taught us that it is not as a disease but only an inherited condition that creates high risks for catching the CF- associated diseases that sickened and killed so many children and adolescents.
Richard Pogue and I showed this from analysis of information in the CF Patient Database, which we had developed in Minnesota. That observation confirmed the theory that the best treatments of CF children would be preventive treatments to prevent those acquired illnesses. A remarkable new concept since physicians were taught that treatments are for acquired diseases, except for immunizations.
●Patience was something we observed in Patients and families: Patience, hope and forgiveness from both Patients and parents for the slowness of our caregiver’s program and the strong disagreements and fights between CF therapy approaches. It was a hard lesson for the CF Center Directors to learn a second time within a decade. But we did! The Medical Advisory Group of the CF Foundation accepted my suggestion to create a Cystic Fibrosis Cooperative Treatment Committee, which I led for eight years, to organize groups of CF Centers to work together to solve medical problems that required more Patients than existed in any one center. This has evolved into the CF Foundation’s Therapeutic Development Network. The 1970s
●The increasing numbers of teenagers in each CF Center and visible numbers of young adults with CF nationwide became our next teachers. They looked at death and experimented, with impatience and courage, to develop plans for living despite their expected short survival. Some worked to beat their own plans. Others burnt their candles from both ends so, "Oh!" parents and "Oh!" CF caregivers they shone a "lovely light".
They taught us both impatience and courage. They looked at death and experimented with individual and different plans. They personally showed us how they could count their days and how they would plan to use those days. They taught the need of a plan, the need for dedication to the goal of the plan, of independence and determination. They may not always have shown wisdom … but how many teenagers do? They also taught us, when all else is gone, the powers of trust, faith and above all hope.
●The `70s were a real turmoil for CF progress with individual reports of a few adults with CF. Along with these reports came the first papers from data in the CF Patient Database that soon, if not right then, physicians in Internal Medicine needed to become part of each CF Center. The first internist trained to be a CF physician was John Hadden, MD, who learned his skills at the Minnesota CF Center. He later moved to The Sloan- Kettering Foundation in New York City and eventually left Academic research for medical business research. A new thought 20 years too soon. Since the 1980s
●This decade brought Bob Beall to the Cystic Fibrosis Foundation with a new vision that the CF Chapter should direct fund raising exclusively to the CFF so that the money could be used to attract new researchers to solve the problems of CF and to attract new funds from government, industry and other foundations. He is succeeding beautifully. Under his leadership we have seen the number of researchers and dollars from other sources increase 100 fold. The CF caregiver meetings have increased from one-day meetings of less than 100 to 5 day meetings of thousands.
●Along with Bob Beall, CF has taught us that the Patients, the families and the basic scientists and the CF Foundation were, with the CF caregivers, the full extension and composition of what we began to call the CF team. Now that CF TEAM is a worldwide team with North Americans and the Europeans attending each other’s annual CF conferences.
●This collaboration led the way to one of the greatest gifts we have had from CF: the working together to identify the CFTR gene. This, the first gene of any disease to be identified, was accomplished in 1988. Two other wonderful discoveries followed: the first that the CFTR gene can have many mutations and that each mutation carries a different risk of catching CF-associated diseases. Since then the genes causing many other diseases have been found, also with many gene mutations but not as many as occur in the CF gene. Another next direct result of the CF gene discovery has been that other genes exist, some of which can worsen and some of which can diminish the risks caused by each CFTR mutation.
●In the `80s the CF families taught us that medical research could be sponsored in two kinds of ways and in two kinds of locations. I will tell of only two of the four possible examples of ways that CF Patients and families have shown us how to help both local CF Centers and the national CF Foundation’s research projects.
LOCAL CF RESEARCH: At our Minnesota CF Centers there was great interest in developing a better way to perform airway clearance. Our vest project was funded entirely by donations from CF families. Umberto Marzotto (Venice, Italy) funded the development of one vest for his daughter Annalisa. Her brother Matteo Marzotto and her husband Riccardo Boatto, both who flew from Italy to attend the CF Gala, helped in designing the early models. The success of the vest for Annalisa inspired personal donations from friends, families and relatives of other CF Patients, which enabled us to build the 16 machines used for the first two-year study of this better way to clean the airways. There is no better example of how the combined gifts of CF families can create the possibility for a new idea, too new and too small for CF Foundation or Federal NIH grants, to become a reality. Now over 20,000 Patients with CF are routinely using this Minnesota invention that came from small gifts from families who had little to spare. The high frequency chest compression vest is a gift from Minnesota CF families to the world.
●CF FOUNDATION RESEARCH: I have already mentioned the discovery of the CF gene but I did not mention the almost 100 Minnesota CF families who donated blood and family histories and so provided one of the largest single blocks of family blood and data that went into the CFTR gene discovery. All this CF community effort was done with only the hope that someday some help might come. This discovery was possible only because CF Patients, parents, families and friends gave time, energy and money to the CF Foundation for the big research projects and the recruitment of other foundations, the NIH and many pharmaceutical and other industries to support and undertake expensive projects that develop from seed projects in the CF Centers.
●A very important contribution of the `90s was that pediatric and internal medicine specialists began to talk to and work with each other and to collaborate with other medical specialists to treat and to strive for a cure for CF. That Minnesota has been in the forefront this kind of continuity of effort is an important reason why the U of M Center is one of the largest and possibly the world’s best CF Center.
●Another local gift to the center from you, CF Patients and families, has been your willingness to help us learn through participation in research projects and through, after death, autopsies. In my experience you have never turned down these opportunities. Such faith in our Center’s efforts to control and to search for a cure for CF have been as important as your personal contributions of money to help CF research move to higher and broader knowledge about CF as a condition with increased risks of catching diseases and of the CF-associated diseases as the problems that kill.
●The gift of a cure for these problems will continue to be become closer because of your selfless aid in these three ways. ●Another good thing is the courage of CF Patients and families that inspires everyone who works with them.
● Fr. Gregory Tolaas represented this courage and vision most clearly. He died this year at age 47, which coincidently is the life table survival for CF Patients at the Minnesota CF Center. In no other way has Greg ever been average. Doug Grow in his December 15, 2002 Star-Tribune column quotes Greg, "I have no fear of death, … I have increasing knowledge --- I guess you call that faith --- that death is a word that only defines what happens to the body. But what it is, is a transition to the bigger, the lovelier, a sense of communion with those who have come before. We’re more infinite than finite." Greg was committed to the end; we mourn our loss; and we gain strength for our continuing work to appreciate the good things about CF even as we are more resolved to solve the problems of CF
●Just as you and other CF Patients and families are in this fight to find better treatments and finally a cure for CF, so the CF Center staff is clearly committed in this to end. Consider their dedication and the number who have dedicated much of their academic life to that end. Karen Reigstad and Evelyn McKee started to work with me in the CF clinic, 34 and 28 years ago. Now they work in the CF Center office daily answering CF patient and physicians calls, and calling back on many of the calls. They come to work early and stay overtime (unpaid) and answer every phone call and when necessary find one of the CF Center Doctors for consultation.
Therapists and Technicians who have served CF Patients for similar times include Ione Brown, Manager of the CF Pulmonary Laboratory and Sweat Testing; Kathy Schissel, Dietician, 27 years; Mary Jo McCracken, who developed the CF Education Day which has been copied around the country and the world, 24 years; Ann Hazelwood Respiratory Therapist, 21 years; Mary Jo Link and Susan Buffie, Clinic Nurses, 34 years. Bonnie Holme who supports everyone in the CF Center Staff in their work to help you has been doing this for 15 years. Jackie Zirbes, Nurse Practitioner and coordinator for lung transplantation, for 11 years. And there have been two who have spent their entire University careers working towards the cure of CF: Leland Hansen, who among other things worked with me to develop the vest therapy for 39 years, and Sue Severson who supervised research projects for 20 years.
Physicians too have spent long times working with CF Patients: Drs. Warren Regelmann for 27, Sara Jane Schwarzenberg (Gastroenterology) for 25, Toni Moran (Endocrinology and Diabetes) for 12, Frank Rimell (ENT) for 15, and Preston Williams (Obstetrics) for 28 years.
●A new era for the CF Center started four years ago when Dr. Carlos Milla, who had worked with me for six years before, became my successor as CF Center Director. Carlos Milla, with wonderful energy has taken on the administration of the CF Center and is leading the CF Center team to national and international recognition.
Since then many new faces have appeared in the CF Center roster. The minds behind these faces are equally dedicated to the ‘good things about CF’ that you, CF Patients and CF families, have been teaching us. The Internal Medicine group includes: Dr. Jordan Dunitz, Director of the Center’s adult program assisted by Dr. Joanne Billings and Dr. Bob Kempainen and Nurse Sandy McAllister. Other newcomers include Dr. James Phillips, Pediatrics; Gretchen Kirvida, Social Worker; Brooke Noren, coordinator for clinical research projects, Teresa Kleinschmidt, Clinical Psychologist and Family Therapist and Josh Olson whose vital secretarial service supports the health care of all CF Center Patients. At the end of this review of good things about CF, I thank the CF Patients and their families for the inspiration that they, their lives and efforts, have given to me over these 42 years.
●And special thanks to:
Lee Hansen, whose wonderful visions of what might be, has been the key to the many developments that have been made in Minnesota: three new kinds of sweat tests, the stereo-stethoscope, several novel analytical tests for vitamins, and three generations of waveform devices for high frequency chest compression VESTS. Also to Kathy, his wife, who must have had many a questioning thought about the crazy things that Warwick thinks of.
Annalisa Marzotto, her husband Ricardo Boatto, her brother Matteo and her father Umberto. Annalisa, who was the beneficiary of Umberto’s funding the development of her vest, along with Ricardo and Matteo made many critical observations and suggestions that made our efforts to develop the vest successful. After Annalisa’s death Umberto donated $1,000,000 to the Medical Foundation to create a Chair in Annalisa’s name to support my research directed towards care of CF Patients. Ricardo and Matteo flew from Italy to Minnesota to celebrate this award with me.
And the women who made it possible for me to do my work with CF deserve immense thanks. My wife Henrietta and our daughters, Marion, who flew here from Washington DC, and Anne, who flew here from Germany have been forgiving when I have spent their time to be at the University Hospital caring for CF Patients who then might have more time with their families.
A special thanks to Bob Beall who spends days and weeks (adding up to months) away from his family so that CF Patients around the world will be able to spend more time with their families. Finally, to the Volunteers, the staffs of the Minnesota CF Chapter and the Minnesota CF Center, to all who came to the 2003 Breath of Life Gala in support of the ‘Angela Warner Friend of the Foundation Award for Lifetime Achievement’, especially to families and Patients who have made great financial sacrifices to come, a special THANK YOU!
As time continues, I will carry on seeing Patients and doing research in four areas.
1) Repairing the CF gene mutations. a. We have organized a team of researchers from two Universities to a practical way to repair each kind CFTR mutation in the nuclei of enough of the cells of the so that the tissues and organs in a CF patient’s body will have the equivalent function of a CFTR gene mutation carrier, i.e., will be as healthy as any other normal person. b. Working with a team of U of M researchers from the Vet School, Med School and College of Agriculture to create a pig with the Δ F508 mutation so that we may: a. Treat pigs with CF to learn better ways to treat humans with CF b. Use the technology in to repair the CFTR mutation in the pig’s cells and pave the way to repair the CF mutations in our CF Patients. c. Developing a simple and fast sweat test the may make newborn diagnosis of CF a reality so the repair of each CF baby’s gene mutations can be done before significant CF associated diseases are acquired. 2) Studying high frequency chest compression (HFCC) with graduate students: a. Evaluating a new waveform device that may come to market at an affordable price. b. Studying three waveforms usable for HFCC so that all may clean the airways more effectively with the long term goal that the lung infections can be removed effectively with the hope that damaged airways can recover normal function. 3) Creating a worldwide study of aging in CF Patients. We are certain that Geriatric CF will be a future specialty and that it is time to add this specialty to our CF Center program. In our 2002 preliminary survey of 12 countries we have found 1865 adults ages 40 and older: eight in the 70s and one age 82. 4) Completing the development of two new pulmonary function tests that will be a. The first to make home monitoring of lung function easy, exact and inexpensive. b. The second to precisely detect and early detection of worsening disease changes or of improvements from treatments. This new test will require only a minute of quiet breathing instead of the maximum efforts of our usual pulmonary function tests.Warren J. Warwick, M.D. Professor of Pediatrics and of Biomedical Sciences and Medical Physics Annalisa Marzotto Professor for Cystic Fibrosis Patient Care MMC 742 Mayo Building 420 Delaware Street SE Minneapolis, MN 55455 Email: warwi001@umn.edu Phone: 612-624-7175 Fax: 612-624-0696